Autoimmune Polyendocrine Syndrome: Types and Symptoms
Reviewed by the LabReadAI medical team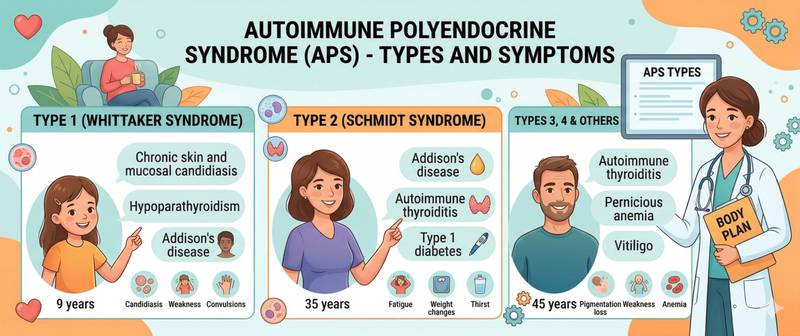
Hashimoto's thyroiditis and type 1 diabetes in the same patient. Adrenal insufficiency alongside hypothyroidism and vitiligo. Chronic candidiasis, hypoparathyroidism and Addison's disease from childhood. These are not coincidences — they are manifestations of autoimmune polyendocrine syndrome, in which the immune system attacks several endocrine glands in succession. Understanding this syndrome matters because when one autoimmune endocrine disease is confirmed, the others must be actively sought.
What Is Autoimmune Polyendocrine Syndrome
Autoimmune polyendocrine syndrome (APS, polyglandular autoimmune syndrome, PAS) is a clinical condition in which a single patient develops two or more autoimmune diseases affecting endocrine glands and/or other organs with tissue-specific autoantibodies, either simultaneously or sequentially.
APS is rooted in a breakdown of immunological tolerance — the immune system's ability to ignore the body's own tissues. In APS this mechanism fails at multiple points simultaneously. This is partly explained by shared genetic factors: many autoimmune endocrine diseases are associated with the same HLA variants (particularly HLA-DR3 and HLA-DR4), creating a common genetic substrate for multiple autoimmune processes in a predisposed individual.
Among endocrinologists, the rule is clear: when any autoimmune endocrine disease is diagnosed, don't wait for symptoms of the next — screen actively. This rule saves lives, especially where adrenal insufficiency is concerned: unrecognised adrenal insufficiency can cause a life-threatening crisis under the stress of intercurrent illness or surgery.
Classification: Types of Autoimmune Polyendocrine Syndrome
Several classification systems exist for APS. The most widely used divides the condition into four types, each with characteristic components and genetic features.
APS-1 (APECED, Whitaker syndrome) — a rare hereditary condition with autosomal recessive inheritance caused by mutations in the AIRE gene (autoimmune regulator). AIRE ensures that immune cells in the thymus "learn" to tolerate the body's own tissues — without it, self-tolerance fails. Disease presents in childhood. Classic triad:
- chronic mucocutaneous candidiasis (usually the first sign, appearing at age 3–5);
- hypoparathyroidism (seizures from hypocalcaemia);
- Addison's disease (adrenal insufficiency).
Additional components of APS-1: hypogonadism, atrophic gastritis, coeliac disease, alopecia, vitiligo, chronic hepatitis.
APS-2 (Schmidt syndrome) — the most common type, occurring in adults and significantly more often in women. The obligatory component is Addison's disease, to which one or more of the following are added:
- Hashimoto's thyroiditis or Graves' disease;
- type 1 diabetes mellitus.
APS-3 — autoimmune thyroid disease (Hashimoto's or Graves') combined with another autoimmune condition but without Addison's disease:
- APS-3a: thyroid disease + type 1 diabetes;
- APS-3b: thyroid disease + atrophic gastritis / pernicious anaemia;
- APS-3c: thyroid disease + vitiligo or alopecia;
- APS-3d: thyroid disease + other autoimmune condition.
APS-4 — a combination of two or more autoimmune endocrine diseases that does not fit types 1–3.
In practice, APS-3a (Hashimoto's + type 1 diabetes) and APS-2 are encountered most frequently. Some specialists simplify the classification to two groups: APS-1 (monogenic, paediatric) and APS-2 (polygenic, adult).
Autoimmune Polyendocrine Syndrome Symptoms: How Multiple Diseases Present
The symptoms of APS are the sum of the manifestations of each constituent disease, but their combination frequently produces atypical clinical pictures that complicate diagnosis.
Profound fatigue and weakness — in APS-2, the combination of hypothyroidism and adrenal insufficiency produces a degree of asthenia substantially greater than either condition alone. Patients often spend years treated for "fatigue" without the adrenal component being identified.
Hypoglycaemia during insulin therapy — in APS-2 combining type 1 diabetes with adrenal insufficiency, previously adequate insulin doses begin causing dangerous hypoglycaemic episodes because cortisol — a counter-regulatory hormone — is deficient. This is an important clinical trap.
Impaired response to levothyroxine — in a patient with hypothyroidism starting thyroid hormone replacement, undiagnosed adrenal insufficiency may be unmasked: thyroxine accelerates cortisol metabolism and the inadequate adrenal reserve becomes symptomatic. Starting thyroid hormone replacement without first excluding adrenal insufficiency is a classic error that can precipitate a crisis.
Seizures and paraesthesiae in APS-1 — a consequence of hypocalcaemia from hypoparathyroidism. Blood calcium and parathyroid hormone should be checked in all APS-1 patients regardless of symptoms.
Skin manifestations — vitiligo (skin depigmentation) and alopecia (hair loss) frequently accompany any type of APS and serve as visible markers of ongoing autoimmune activity in the individual patient.
Polyendocrine Syndrome Diagnosis and Screening After the First Autoimmune Disease
The key diagnostic principle in APS: don't wait for symptoms — search actively. Once any autoimmune endocrine disease is confirmed, regular screening for related conditions is mandatory. Screening frequency depends on APS type and individual risk — typically annually.
In Hashimoto's thyroiditis or hypothyroidism — screen for:
- 21-hydroxylase antibodies and morning cortisol — to exclude subclinical adrenal insufficiency;
- fasting glucose and anti-GAD / ICA antibodies — to exclude type 1 diabetes;
- vitamin B12 and intrinsic factor antibodies — to exclude pernicious anaemia.
In type 1 diabetes mellitus — screen for:
- TSH and anti-TPO antibodies — thyroiditis is found in 15–30% of patients with type 1 diabetes;
- 21-hydroxylase antibodies — Addison's disease occurs in 0.5% of type 1 diabetes patients, but risk is many-fold higher when antibodies are present;
- anti-transglutaminase antibodies — coeliac disease occurs in 5–10% of type 1 diabetes patients.
In Addison's disease — the broadest screening is warranted given the high comorbidity: TSH + anti-TPO, fasting glucose, anti-parietal cell and anti-intrinsic factor antibodies, anti-17α-hydroxylase antibodies (for autoimmune hypogonadism).
Instrumental workup:
- bone densitometry — bone density is reduced in hypoparathyroidism (APS-1) and adrenal insufficiency;
- complete blood count — to detect anaemia in pernicious anaemia or coeliac disease.
Genetic Aspects: Which Relatives Need Testing
For APS-1 (AIRE mutation) — autosomal recessive inheritance. Testing of siblings is mandatory, particularly when there is a history of recurrent candidiasis or childhood seizures.
For APS-2 and APS-3 — polygenic predisposition. No single candidate gene has been identified, so genetic screening of relatives is not standardised. However, first-degree relatives of APS-2 patients are advised to have periodic TSH and fasting glucose measurements as a minimum monitoring approach.
In women with PCOS and cycle disturbances when an autoimmune endocrine disease is already established, autoimmune oophoritis as a component of APS should be excluded — particularly when premature ovarian insufficiency develops.
Treatment of Autoimmune Polyendocrine Syndrome
Treatment of APS is the treatment of each of its constituent diseases. No unified pathogenetic therapy capable of halting the autoimmune process in multiple glands simultaneously currently exists.
Priority of life-saving replacement therapy. In APS-2 combining hypothyroidism and Addison's disease: glucocorticoid replacement (adrenal insufficiency treatment) is initiated first, and levothyroxine is added only after stabilisation. The reverse order is dangerous.
Dose monitoring. With type 1 diabetes combined with adrenal insufficiency, insulin doses typically require downward adjustment once glucocorticoid therapy is started, and upward adjustment if glucocorticoid doses are ever reduced. Glycaemic management becomes considerably more complex.
Hypoparathyroidism in APS-1 is treated with calcium supplements and active vitamin D analogues (alfacalcidol, calcitriol). Unlike ordinary hypoparathyroidism, recombinant parathyroid hormone is recommended in severe APS-1 cases.
Immunosuppression. In APS-1 with severe cutaneous or systemic manifestations, tacrolimus or other immunosuppressants are used. In other APS types, immunosuppression has limited indications and generally does not alter the endocrine component of the disease.
All patients with APS are advised to wear a medical alert bracelet stating their adrenal insufficiency diagnosis, and to learn emergency hydrocortisone self-injection technique — particularly given the risk of acute crisis during intercurrent illness.
When to See an Endocrinologist
See an endocrinologist for APS screening when:
- you have been diagnosed with Hashimoto's thyroiditis, type 1 diabetes or Addison's disease — annual screening for related autoimmune conditions is recommended;
- a child has recurrent mucocutaneous candidiasis, seizure episodes or unexplained developmental delay — exclude APS-1;
- a patient on insulin begins experiencing unexpectedly severe hypoglycaemia — adrenal insufficiency must be excluded;
- multiple first-degree relatives have different autoimmune endocrine diseases — screening is warranted.
Screening for vitamin B12 deficiency is important in all APS types: atrophic gastritis with pernicious anaemia frequently accompanies autoimmune thyroid disease and often goes unrecognised. Do not interpret symptoms independently — consult a physician.
This content is for informational purposes only and does not replace professional medical advice.
For informational purposes only
This article is for informational purposes only and does not constitute medical advice, diagnosis, or treatment. Please consult a healthcare professional for medical guidance.